Spinal muscular atrophy in children

Spinal muscular atrophy is a severe pathology that often makes simple actions, such as walking, sitting, inaccessible to a child. The baby may even be deprived of such a natural opportunity as independent breathing. It is very difficult to make predictions for this pathology, after all, neither special treatment nor prophylaxis exists, but everything depends on the form of the affliction and factors, which medicine can not find an explanation.
What is it?
Speaking of spinal muscular atrophy, imply not one specific disease, but a whole group of diseases under the general abbreviation CMA. All of them are hereditary and are associated with the degeneration of the nerve cells of the spinal cord, which are responsible for motor functions.
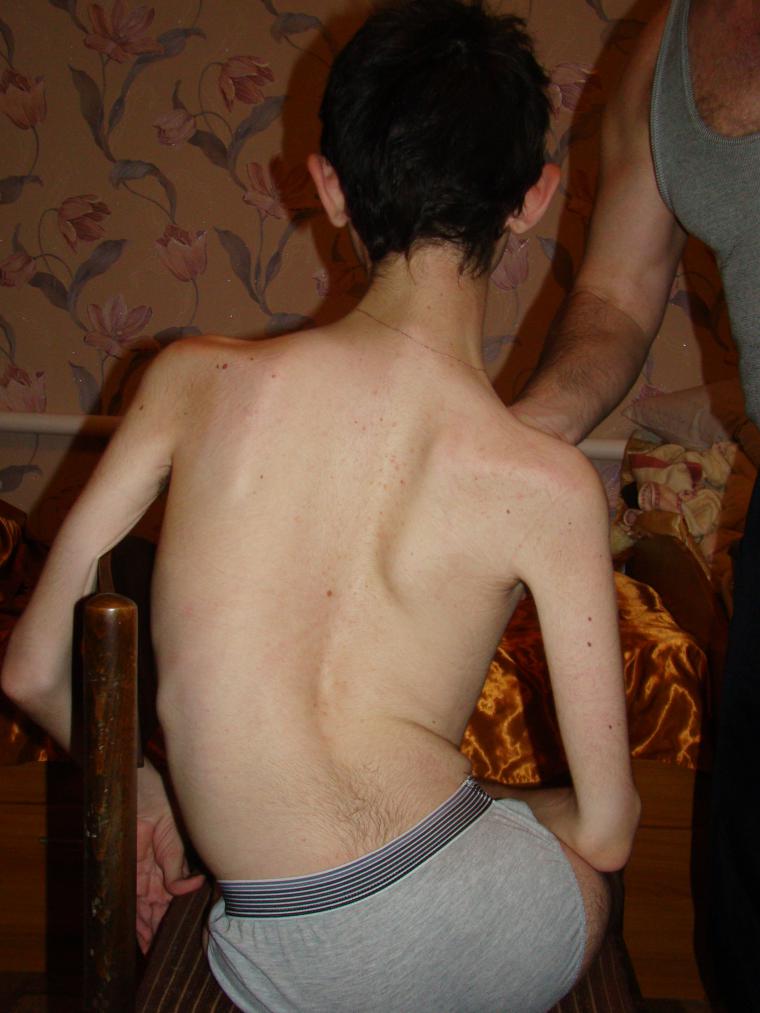
Among genetic pathologies in children spinal muscular atrophies occupy leading places in terms of frequency of spread. And about one in 6 thousand children is born with such a terrible diagnosis. In 50% of cases, children do not live to the age of two and die. The rest of the life is a disability.

The problem, according to geneticists, is much broader than it may seem from the statistics given.
The disease develops due to the mutation of certain genes, and one of them is SMN1, which is considered the main “culprit” of pathology, in every modified form, every fiftyth inhabitant of the planet is present in recession. This means that healthy parents, who do not even realize that they are recessive carriers of the mutated gene, may well have a baby with spinal muscular atrophy.
The group of diseases was first described in the 19th century by Guido Verding, whose name was later named one of the children's varieties of AGR.
Classification
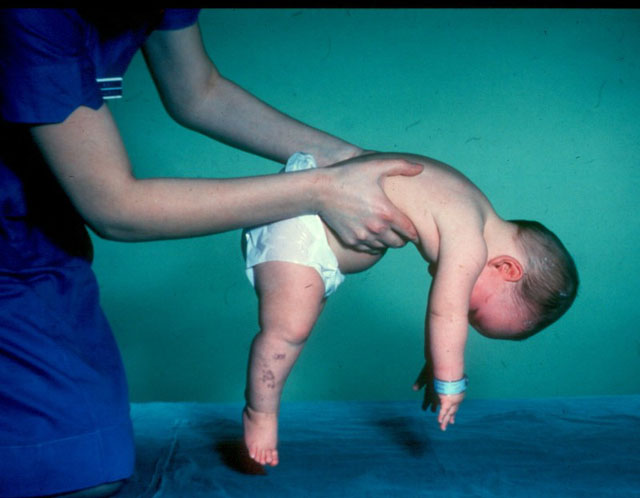
The most common form of SMA in children is proximal. It is represented by several types of disease, not all of which become apparent immediately after the birth of the child.
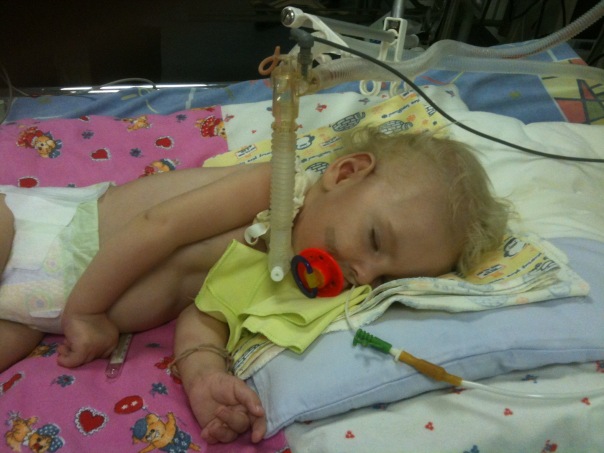
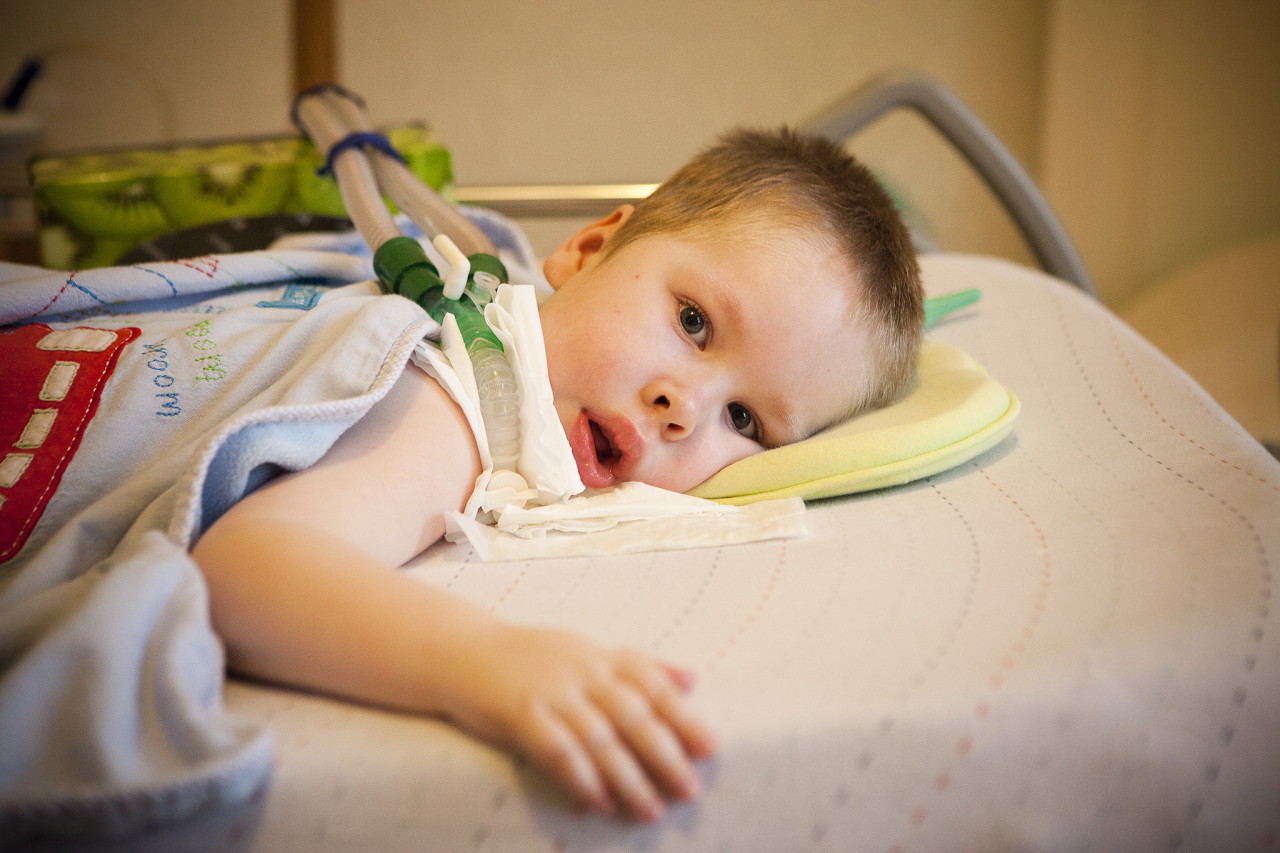
- Verding-Hoffman's disease - CMA type 1, severe infant diseasewhich manifests itself in the first six months of the child’s life. Forecasts with her most unfavorable, most patients die. A child with type 1 SMA can neither stand nor sit or roll over on its own. Many newborns have impaired sucking and swallowing reflexes. Often there is no possibility of spontaneous breathing or breathing is difficult.
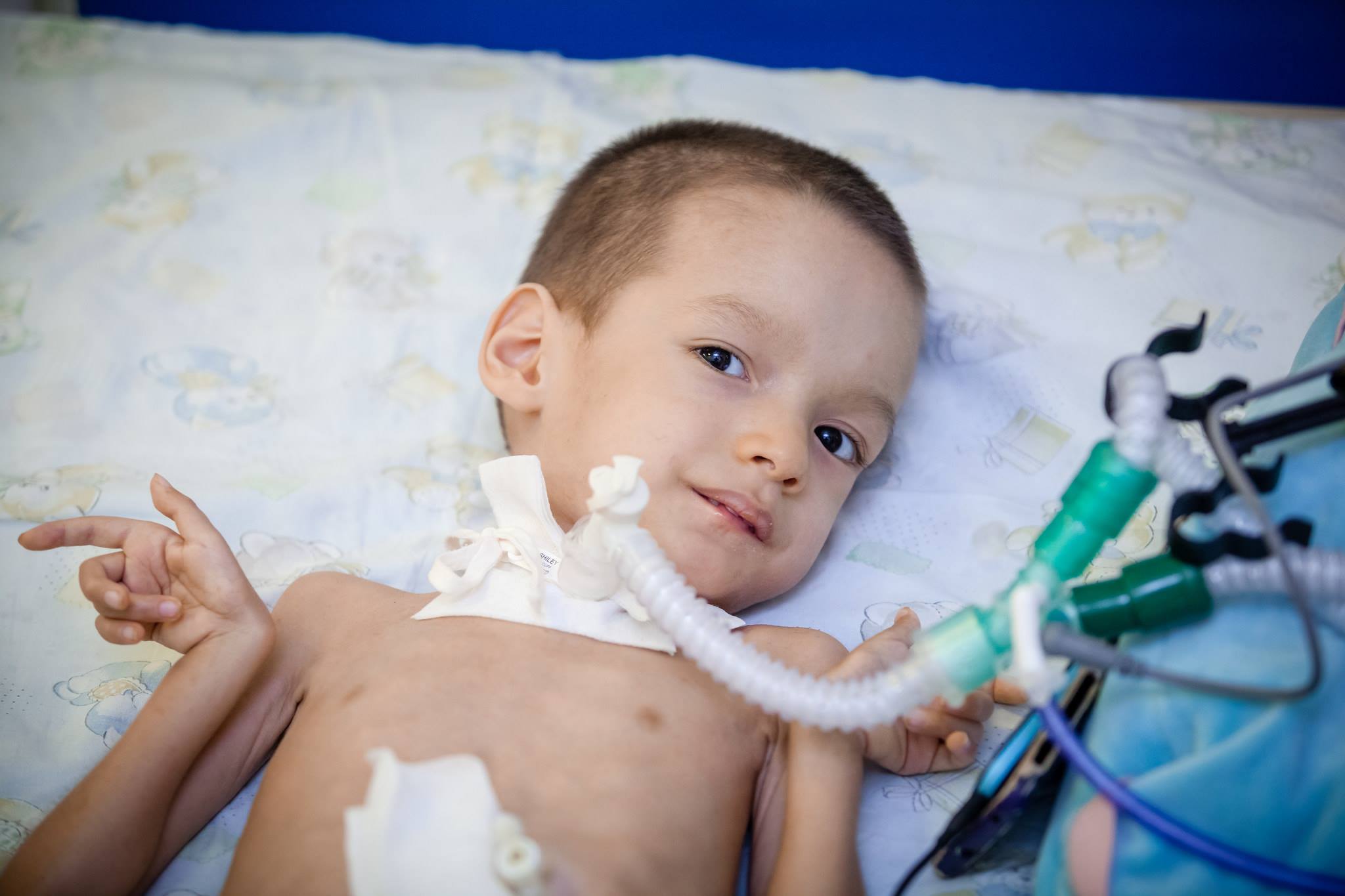
- Atrophy Dubovitsa - CMA type 2, late infant. It usually occurs between the ages of six months and a year and a half and later. The child cannot walk, stand, but is able to sit, food is not disturbed, he copes with the task of swallowing, sucking. How long the baby will live depends on the state of the respiratory muscles.
- Atrophy of Kugelberg-Welander - CMA 3 types, infantile. Usually found in the age of one and a half years, usually in two years. Prognostically more favorable form. Small patients can stand, sit, move, but experience great weakness, and therefore in most cases need a wheelchair, without which their normal vital activity is difficult for them.
- Kennedy atrophy - CMA type 4, bulbospinalnaya. Usually considered an adult form, but occasionally detected in children after 15 years. Influence of life is rarely affected, weakening of muscles occurs slowly, gradually, a person who led a normal life and considered himself healthy, eventually becomes disabled and loses the ability to move independently.


More or less known first-hand atrophy of Duchenne and atrophy of Vulpian is SMA “adults,” the first is usually detected after 18 years, and the second after 20 years.
In children, not only isolated forms of SMA are recorded, when nothing bothers except muscle dystrophy, but also combined forms, when spinal atrophy is not the only diagnosis and the child has other genetic or congenital problems, such as heart and vascular defects, oligophrenia.
The reasons
As already mentioned, we are talking about a genetic disease, and therefore the reasons for its occurrence is the field of searches for geneticists. The child inherits one of the recessive genes on the fifth chromosome (these may be the SMN, NAIP, H4F5, BTF2p44 genes).
The probability of transmitting such a gene to a progeny from a carrier is high - 25%. If both mom and dad are hidden carriers of the mutated gene, then the probability of AGR in a child is 50%. The affected abnormal gene prevents normal SMN protein production and the nerve cells responsible for the motor functions of the muscles in the spinal cord gradually begin to die. The process of their death continues even after the baby is born.


Manifestations
Symptoms depend on the type of illness. Since we consider only four types of children, it should be noted that muscular weakness and muscular atrophy are characteristic of all. Otherwise, each type has its own clinical picture and distinctive features.
- CMA Type 1 (Verding-Hoffman Atrophy) available for detection even during pregnancy. The doctor may suspect the disease in the fetus with very sluggish stirring. But to confirm the diagnosis at the stage of carrying the child is difficult, it usually happens after giving birth. A kid with such atrophy cannot hold the head himself, toss from side to side, he does not sit down. He almost always lies on the back, his posture is relaxed, he does not raise his legs, does not bring them together, does not put his palms together. At a very early stage, there can be huge problems in order to feed the child, because he swallows it turns out very badly or does not work. Most of the children die before the age of two. Some manage to live to seven or eight years, but atrophy only increases. Usually the death occurs due to the insufficiency of the heart, lungs, digestive organs.
- CMA 2 type (atrophy Dubovitsa) at birth, it is usually not detected, because the child is able to breathe, swallow food, and only after six months does the progress of muscle atrophy become apparent. If the first symptoms occur at the age when the child has already learned to stand in the crib, then a cutting sign of the legs, an unreasonable drop of the crumbs can be a bright sign. Gradually, it becomes difficult to swallow. Over time, the child begins to need a wheelchair.
- CMA 3 types (amyotrophy of Kügelberg-Welander) may show up at any age after 2 years of age. A child who normally grew and developed gradually begins to complain of weakness, usually in the shoulder area, forearms. As it progresses, it becomes difficult for him to run, walk stairs, squat. It all depends on the care - some retain the ability to move independently for many years.
- CMA type 4 (Kennedy atrophy) is found only in male patients, since it is considered linked to the sex chromosome X. The first signs are weakness in the region of the thigh muscles, the cranial nerves are gradually affected. The disease progresses slowly.

Treatment
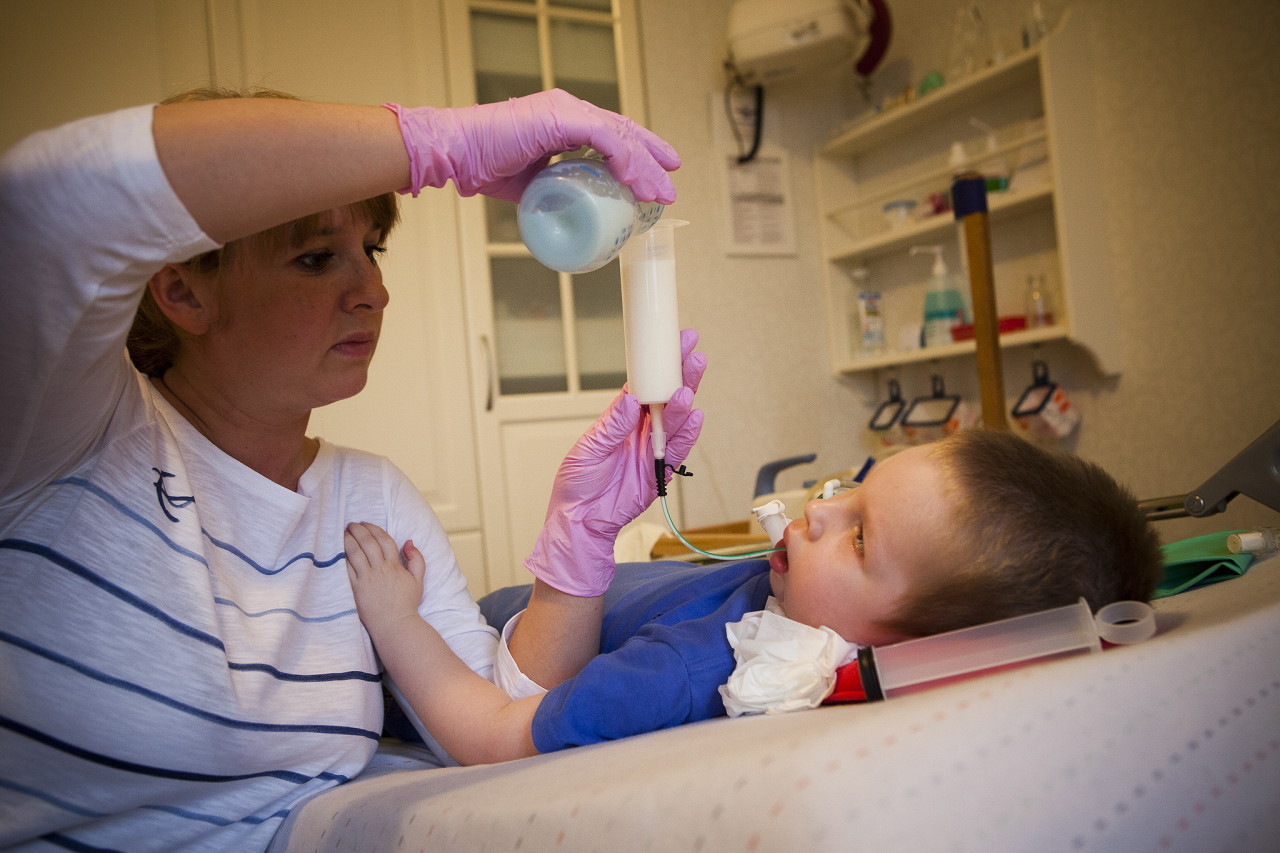
Unfortunately, today medical science cannot offer methods and means for the treatment of SMA. There are no such methods. To maintain the functions of the body and maximize the period until the child can move himself, medications such as Prozerin, Oksazil. They reduce the activity of an enzyme capable of cleaving acetylcholine, which transmits an excitation pulse through the fibers of the nervous system.
Also recommended systematic use of drugs that increase energy metabolism at the cellular level, group B vitamins, nootropic drugs, as well as potassium and nicotinic acid preparations.
A child with SMA high protein diet shownBut recent studies have shown that the role of the diet is somewhat exaggerated - there is no evidence that a high content of protein in food at least in some way affects the rate of progression of the disease.
But with the calories should be more careful - due to reduced muscle activity, the child can quickly gain extra pounds.
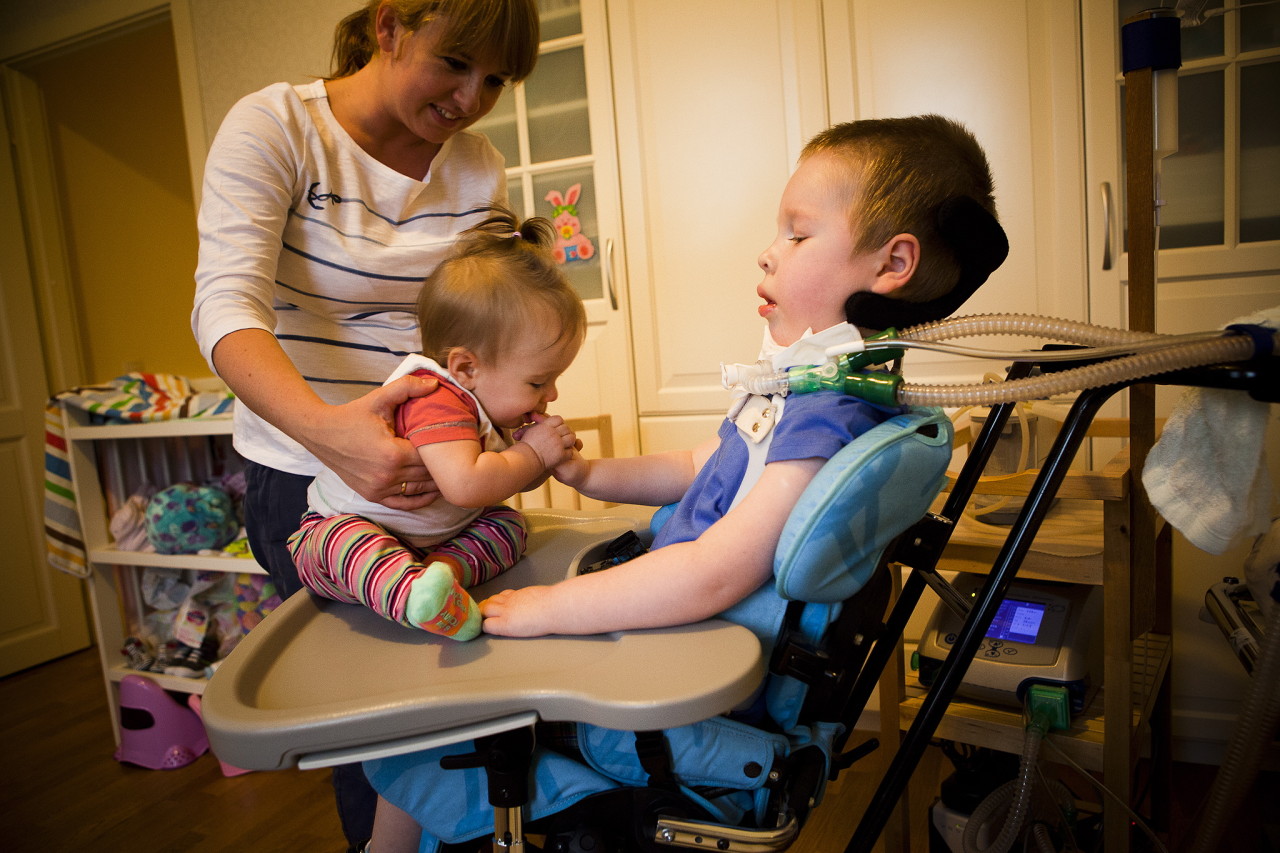
Help extend the period of more or less fulfilling life will help therapeutic massage, UHF, electrophoresis, breathing exercises to maintain respiratory muscles, swimming. Wearing supporting spinal and thoracic orthopedic appliances is recommended.

More information about the disease tells a specialist in the video below.